Abstract and Introduction
Abstract
The loss of functional von Hippel-Lindau (VHL) tumor suppressor gene is associated with the development of clear-cell renal cell carcinoma (CC-RCC). Recently, VHL was shown to promote the transcription of E-cadherin, an adhesion molecule whose expression is inversely correlated with the aggressive phenotype of numerous epithelial cancers. Here, we performed immunohistochemistry on CC-RCC tissue microarrays to determine the prognostic value of E-cadherin and VHL with respect to Fuhrman grade and clinical prognosis. Low Fuhrman grade and good prognosis associated with positive VHL and E-cadherin immunoreactivity, whereas poor prognosis and high-grade tumors associated with a lack of E-cadherin and lower frequency of VHL staining. A significant portion of CC-RCC with positive VHL immunostaining correlated with nuclear localization of C-terminally cleaved E-cadherin. DNA sequencing revealed in a majority of nuclear E-cadherin-positive CC-RCC, subtle point mutations, deletions and insertions in VHL. Furthermore, nuclear E-cadherin was not observed in chromophobe or papillary RCC, as well as matched normal kidney tissue. In addition, nuclear E-cadherin localization was recapitulated in CC-RCC xenografts devoid of functional VHL or reconstituted with synthetic mutant VHL grown in SCID mice. These findings provide the first evidence of aberrant nuclear localization of E-cadherin in CC-RCC harboring VHL mutations, and suggest potential prognostic value of VHL and E-cadherin in CC-RCC.
Introduction
Renal cell carcinoma (RCC) accounts for approximately 3% of all adult malignancies, with the clear-cell type (CC-RCC) comprising 80% of RCC.[1,2] Approximately 25-30% of CC-RCC patients have metastatic disease at diagnosis, and 20-30% of patients with clinically localized CC-RCC develop metastasis post-nephrectomy.[3] Recent advances in the use of antiangiogenesis-targeted agents appear to have revolutionized treatment.[4,5,6,7] Currently, a prediction of patient survival is based on traditional clinical parameters, including tumor size and Fuhrman nuclear grade.[8] However, the emerging understanding of the molecular pathways implicated in CC-RCC genesis and progression is providing previously unappreciated markers, which may serve as additional or better prognostic indicators of CC-RCC.
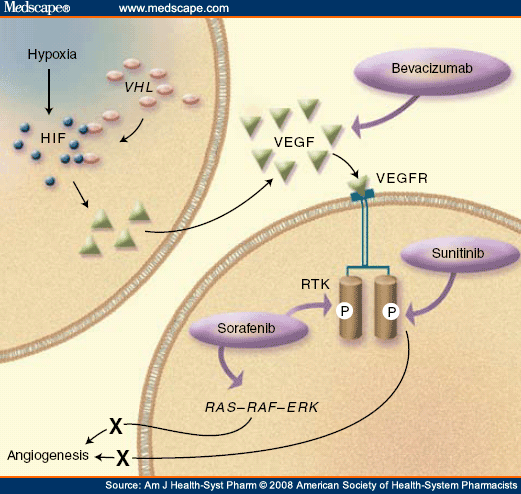
The principal cause of sporadic CC-RCC and familial von Hippel-Lindau (VHL) disease-associated CC-RCC is the inactivating mutations of VHL. Although VHL patients also develop tumors in other organs including the central nervous system, retina and the adrenal gland, CC-RCC remains to be the leading cause of morbidity and death for VHL patients.[9] The most well-established function of VHL is its role in the oxygen-dependent negative regulation of hypoxia-inducible factor (HIF). VHL is a substrate-conferring component of an E3 ubiquitin ligase ECV (elongins/Cul2/VHL) that polyubiquitylates the catalytic α-subunit of HIF that has undergone hydroxylation on conserved prolyl residues within the oxygen-dependent degradation (ODD) domain. Prolyl hydroxylation is mediated by a class of prolyl hydroxylases (PHD1-3) in an oxygen-dependent manner. Thus, under hypoxia or in the absence of a functional VHL, HIFα becomes stabilized and binds to its common and constitutively expressed β-subunit, forming an active HIF transcription factor capable of transactivating numerous hypoxia-inducible genes such as vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT1) and erythropoietin (EPO).[9] These discoveries have confirmed VHL as a critical regulator of the ubiquitous mammalian oxygen-sensing pathway.
HIFα is overexpressed in most tumors including CC-RCC. The re-introduction of VHL into VHL-null CC-RCC abrogates the tumorigenic potential of these cells in a mouse xenograft model.[10,11] Kondo et al[12] have shown that the expression of a non-degradable form of HIF2α was able to restore the tumorigenic phenotype in CC-RCC cells ectopically expressing VHL. Conversely, shRNA-mediated knockdown of HIF2α was sufficient to suppress the tumorigenic capacity of VHL-null CC-RCC cells.[13] Notably, the emerging evidence suggests that VHL mutations affecting HIF regulation were invariably associated with a subtype of VHL disease with a greater propensity of CC-RCC. Although these reports support the notion that deregulated accumulation of HIFα upon the loss of VHL is crucial for the development of CC-RCC, the precise downstream target(s) of HIF that are responsible for renal epithelial oncogenesis have, until recently, remained unclear.
The regulation of adhesive interactions between cells is critical during cell growth and differentiation, and the loss of cell-cell adhesion is frequently associated with tumor progression and metastasis.[14] Recently, VHL was shown to promote the transcription of E-cadherin via HIF-dependent activation of E-cadherin-specific transcriptional repressors.[15,16,17] Thus, a loss of VHL in CC-RCC results in the hyperactivation of HIF that triggers the expression of E-cadherin repressors, which in turn attenuates the expression of E-cadherin.[15,16,17] E-cadherin, a homophilic adhesion molecule associated with catenins that functions as a major component of cell junctions in polarized epithelial cells, is an established tumor suppressor. The graded loss of E-cadherin correlates with the aggressiveness of numerous carcinomas and the worsening of prognosis, while forced expression of E-cadherin suppresses tumor development and invasion in various in vitro and in vivo tumor model systems.[14]
The full-length E-cadherin (120 kDa) can be proteolytically cleaved at a cleavage site near the transmembrane domain, which releases an extracellular N-terminal 80-kDa fragment and generates a 38-kDa C-terminal fragment that can be further processed into a 33-kDa soluble cytosolic fragment.[18] The proteolytic ectodomain release or 'shedding' of E-cadherin is emerging as an important regulatory mechanism and has been suggested to cause rapid changes in cell adhesion, signaling and apoptosis.[19,20,21,22] Furthermore, increased levels of the soluble N-terminal fragment have been associated with several tumors, including prostate,[23] gastric,[24] hepatocellular[24] and bladder cancer and may be of prognostic value.[21] Whether the C-terminal intracellular fragment of E-cadherin has oncogenic roles, for example, via promoting β-catenin-mediated signaling, is unclear.
Here, we examined the prognostic value of VHL and E-cadherin in CC-RCC. While CC-RCC with negative E-cadherin staining exhibited no VHL immunoreactivity as expected, a significant portion of CC-RCC had aberrant nuclear E-cadherin staining despite positive VHL staining. DNA sequencing revealed in a majority of the nuclear E-cadherin-positive CC-RCC, subtle point mutations, deletions and insertions in VHL. The nuclear E-cadherin was not observed in chromophobe or papillary RCC, suggesting this to be specific to CC-RCC. Low Fuhrman grade and better prognosis associated with positive VHL and nuclear E-cadherin immunoreactivity, whereas high-grade tumors associated with a lack of nuclear E-cadherin staining and lower frequency of VHL staining. These findings provide the first evidence of an aberrant nuclear localization of E-cadherin in CC-RCC harboring VHL mutations, and suggest potential prognostic value of VHL and E-cadherin status in CC-RCC.
Materials and Methods
Cell Culture
Human 786-O CC-RCC cells were cultured in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum (Sigma, Saint Louis, MI, USA) at 37°C in a humidified 5% CO2 atmosphere. 786-O subclones stably expressing hemagglutinin (HA)-tagged wild-type VHL30, HA-VHL19(WT), HA-VHL19(MUT) with V84M and P192Q mutations, or empty plasmid were generated as previously described.[25,26]
Antibodies
Anti-HA (12CA5; Roche Applied Science, Basel, Switzerland), anti-TCP1 (Abcam Inc., Cambridge, MA, USA), C-terminal-specific anti-E-cadherin (BD Biosciences, Mississauga, ON, Canada), N-terminal-specific anti-E-cadherin (Vector laboratories, Burlingame, CA, USA), anti-VHL (IG32) (BD Biosciences, Mississauga, ON, Canada), and anti-β-catenin (BD Biosciences, Mississauga, ON, Canada) monoclonal antibodies and anti-Glut1 polyclonal antibodies (Alpha Diagnostic International Inc., San Antonio, TX, USA) were obtained from the companies indicated.
Plasmids
The mammalian expression plasmid pRc-CMV-HA-VHL30 was previously described.[25,26] pRc-CMV-HA-VHL19(WT) was generated from the pRc-CMV-HA-VHL30 plasmid using primer set 5'-CCCAAGCTTATGTATCCATATGATGTTCCAGATTATGCTGAGGCCGGGCGGCCG-3'/5'-GCTCTAGATCAATCTCCCATCCGTTGATGTGC-3'. pRc-CMV-VHL19(MUT), V84M and P192Q, was serendipitously generated from pRc-CMV-HA-VHL30 using the above primers in a PCR reaction. The authenticity of the plasmids was confirmed by direct DNA sequencing.
Immunoprecipitation and Immunoblotting
Cells were lysed in EBC buffer (50 mM Tris (pH 8), 120 mM NaCl, 0.5% NP-40) supplemented with protease and phosphatase inhibitors (Roche, Laval, QC, Canada). Samples were supplemented with the indicated antibody and immobilized on protein A-Sepharose beads (Amersham Biosciences, Uppsala, Sweden). The samples were then washed five times with NETN buffer (20 mM Tris (pH 8), 120 mM NaCl, 1 mM EDTA, 0.5% NP-40), eluted by boiling in sodium dodecyl sulfate (SDS)-containing sample buffer, separated by SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred to a polyvinylidene difluoride membrane (Bio-Rad, Mississauga, ON, Canada). Specific protein bands on Western blots were visualized using the various antibodies indicated. All primary antibodies were diluted in phosphate-buffered saline (PBS) containing 0.1% Tween 20. Horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit or rabbit anti-goat antibody (Pierce, Rockford, IL, USA) was used at 1:20 000 dilution as a secondary antibody, after which enhanced chemiluminescence (PerkinElmer Life Sciences, Woodbridge, ON, Canada) was performed for detection.
Metabolic Labeling
Metabolic labeling was performed as previously described.[27] Briefly, radioisotope labeling was performed by methionine starvation for 45 min, followed by growth in 5 ml of methionine-free Dulbecco's modified Eagle's medium supplemented with [35S]methionine (100 μCi/ml of medium; PerkinElmer Life Sciences, Woodbridge, ON, Canada) and 2% dialyzed fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere for 3 h for the interaction studies and 1 h for half-life analysis.
RNA Extraction
RNA was extracted from cells using RNeasy kit according to the manufacturer's instructions (Qiagen, Mississauga, ON, Canada).
Quantitative Real-time PCR
Quantitative real-time PCR was performed as previously described.[28] Briefly, first-strand cDNA synthesis was performed as follows: 1 μl of oligo(dT)23 primer (Sigma, St Louis, MI, USA) was incubated with 5 μg of RNA and dH2O (total reaction volume was 20 μl) for 10 min at 70°C in a thermal cycler (MJ research, Boston, MA, USA). The mixture was cooled to 4°C, at which time 4 μl of 5 × first-strand reaction buffer, 2 μl of 0.1 M DTT, 1 μl of 10 mM dNTPs and 1 μl Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) were added. cDNA synthesis was performed for 1.5 h at 42°C, followed by 15 min at 70°C in the thermal cycler. Human genomic DNA (Roche, Mannheim, Germany) standards or cDNA equivalent to 20 ng of total RNA were added to the qPCR reaction in a final volume containing 1 × PCR buffer (without MgCl2), 3 mM MgCl2, 0.25 U Platinum Taq DNA polymerase, 0.2 mM dNTPs, 0.3 μl SYBR Green I, 0.2 μl ROX reference dye and 0.5 μM primers (Invitrogen, Carlsbad, CA, USA). Amplification conditions were as follows: 95°C (3 min); 40 cycles of 95°C (10 s), 65°C (15 s), 72°C (20 s), 60°C (15 s) and 95°C (15 s). qPCR was performed using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA). Gene-specific oligonucleotide primers designed using Primer Express (Applied Biosystems) were as follows: GLUT-1 primer set 1 (5'-CACCACCTCACTCCTGTTACTTACCT-3' and 5'-CAAGCATTTCAAAACCATGTTTCTA-3'), GLUT-1 primer set 2 (5'-CTCCCAGCAGCCCTAAGGAT-3' and 5'-ATCTGTCAGGTTTGGAAGTCTCATC-3') and β-actin primer set (5'-GGATCGGCGGCTCCAT-3' and 5'-CATACTCCTGCTTGCTGATCCA-3'). SYBR I fluoresces during each cycle of the qPCR by an amount proportional to the quantity of amplified cDNA (the amplicon) present at that time. The point at which the fluorescent signal is statistically significant above the background is defined as the cycle threshold (Ct). Expression levels of the various transcripts were determined by taking the average Ct value for each cDNA sample performed in triplicate, and measured against a standard plot of Ct values from amplification of serially diluted human genomic DNA standards. Since the Ct value is inversely proportional to the log of the initial copy number, the copy number of an experimental mRNA can be obtained from linear regression of the standard curve. A measure of the fold difference in copy number was determined for each mRNA. Values were normalized to the expression of β-actin mRNA and represent the average value of both GLUT-1 primers in three independent experiments performed in triplicate.
In Vitro Ubiquitination Assay
An in vitro ubiquitination assay was performed as previously described.[29] The [35S]methionine-labeled reticulocyte lysate HA-HIF-1α ODD translation product (4 μl) was incubated in 786-O RCC S100 extracts (100-150 μg) supplemented with 8 μg/μl ubiquitin (Sigma), 100 ng/μl ubiquitin aldehyde (Boston Biochem Inc., Cambridge, MA, USA) and an ATP-regenerating system (20 mM Tris (pH 7.4), 2 mM ATP, 5 mM MgCl2, 40 mM creatine phosphate and 0.5 μg/μl creatine kinase) in a reaction volume of 20-30 μl for 1.5 h at 30°C.
Protease Digestion Assay
Protease sensitivity was assayed as previously described.[30] Briefly, lysates were incubated with chymotrypsin (10 μg/ml) for 10 min on ice. Reactions were stopped by the addition of 1 mM PMSF before immunoprecipitation (IP) and SDS-PAGE.
SCID Mouse Xenograft Assay
Multiple 786-O subclones expressing HA-VHL30, HA-VHL19(MUT) or empty plasmid were grown to ˜ 90% confluence in a humidified 5% CO2 atmosphere at 37°C. Cells were harvested with a solution of 0.25% trypsin and 1 mM EDTA. Cells (1 × 107) in 100 μl of 1 × PBS were injected intramuscularly into the left hind legs of male SCID mice (Charles River Laboratories Inc., Wilmington, MA, USA). Tumor growth was assessed and measured weekly by carefully passing the tumor-bearing leg through a series of holes of decreasing diameter (0.5-mm decrements) in a plastic rod.
Immunohistochemistry
SCID mouse xenograft tumors were fixed in 10% neutral buffered formalin and later embedded in paraffin. Formalin-fixed, paraffin-embedded sections from 19 nephrectomy specimens with CC-RCC were obtained from the Department of Pathology and Laboratory Medicine at The University Health Network (Toronto, ON, Canada). These tissue blocks were used and processed in accordance with a University Health Network Research Ethics Board-approved protocol concerning gene expression in RCC. Tissues were fixed in 10% neutral buffered formalin for 24-36 h. Representative sections of tumor with adjacent non-tumor renal parenchyma, 3-4 mm in thickness, were embedded in paraffin and 5-μm sections were cut and placed on coated slides for light microscopy.
Tumor morphology and classification were assessed using standard hematoxylin and eosin (H&E) staining. The tumors were classified as conventional clear-cell type according to criteria described in the WHO classification of renal tumors.[31] Immunohistochemical staining for VHL was performed manually using a standard avidin-biotin-peroxidase complex method. Briefly, unlabeled anti-HA, anti-Glut1, anti-E-cadherin (BD biosciences, Mississauga, ON, Canada), anti-E-cadherin (Vector laboratories, Burlingame, CA, USA) and anti-β-catenin were used at a 1:2000 dilution, and an overnight incubation following microwave pretreatment for antigen retrieval. The sections were then incubated with a biotinylated secondary antibody (horse anti-mouse IgG, 1:200 dilution) and the avidin-peroxidase complex. The colored reaction was visualized using nova red as the chromogen. The tissue was then mildly counterstained with hematoxylin.
Tissue Microarray
Tissue microarray (TMA) consisting of quadruplicate representative 1.0-mm cores from 56 CC-RCC with various Fuhrman grades (grades 1-4) and stages (pT1-pT4) were used to analyze the correlation between E-cadherin and VHL protein expression. Sections (5 μm) from the CC-RCC TMA were stained with anti-E-cadherin and anti-VHL antibodies as described above. The slides were then scanned using a ScanScope (Aperio Technologies, Vista, CA, USA) and scored by two observers. In order to be considered for evaluation, at least half of the TMA core was required to be present on the slide, with at least 50% of the tissue present being tumor cells. Cores were being scored as either positive or negative. Only tumors with the majority of cores in agreement (3 out of 4 or 2 out of 3) were used for further analysis.
VHL Sequencing
VHL was amplified from patient samples using primers 5'-CAGTAACGAGTTGGCCTAGC-3'/5'-GGATGTGTCCTGCCTCAAG-3' for exon 1, 5'-GGACGGTCTTGATCTCCTGA-3'/5'-GATTGGATACCGTGCCTGAC-3' for exon 2 and 5'-CTGCCACATACATGCACTCA-3'/5'-AAGGAAGGAACCAGTCCTGT-3' for exon 3. PCR reactions were performed using the Qiagen Multiplex Master Mix (Qiagen, Mississauga, ON, Canada) according to manufacturer's instructions. Exons were sequenced with the Cy5/Cy5.5 Dye Primer Cycle Sequencing kit (Amersham, Quebec/Visible Genetics, Toronto) according to the manufacturer's instructions. Each primer mixture contained two primers, labeled with either Cy5 or Cy5.5. Primers used for sequencing were 5'-/5Cy5/-CTAGCCTCGCCTCCGTTA-3'/5'-/5Cy5/-GCCTCAGTTCCCCGTCTG-3' for exon 1, 5'-/5Cy5-GATACCGTGCCTGACATCAG-3' for exon 2 and 5'-/5Cy5/-CAGGTAGTTGTTGGCAAAGC-3' for exon 3. Two primers were used for exon 1 as it is a large exon. The sequences generated were compared with wild-type VHL (GenBank accession number AC_000046, for sequence alterations, using the OpenGene Automated DNA System and Gene Librarian software, version 3.1 (Visible Genetics, Suwanee, GA, USA).
Statistics
Fisher's exact test was used to determine whether positive VHL immunoreactivity and nuclear E-cadherin subcellular localization were interdependent. Two-tailed P-values less than 0.05 were considered significant.